Menu
Menu
Chromosomal conditions are caused by changes in the number or structure of chromosomes. It is estimated that 1 in 150 babies is born with a chromosomal condition, and each child is different.1 Some children with chromosomal disorders may have intellectual disabilities and/or birth defects and some may have no serious problems, depending on which chromosomes are affected and how.
This year we are spreading awareness of Rare Chromosome Disorders and celebrating the unique children and adults with these disorders through our fun quiz. Give it a try and test your knowledge!
Our team at SOPHiA GENETICS continue to support the management of the full spectrum of rare and inherited diseases by advancing data-driven medicine.
1. March of Dimes, Chromosomal conditions. Accessed June 2022.
Bita Khalili is a Senior Algorithm Researcher in our SOPHiA GENETICS Data Science team. She joined the team after completing her PhD in Physics and a post-doctoral research position in Bioinformatics. For the last two years, Bita has been analyzing NGS data at SOPHiA GENETICS and developing copy number variation (CNV) detection modules.
We invite you to spend a few moments with Bita to learn about the challenges associated with CNV detection and how MUSKAT™, the SOPHiA GENETICS' CNV detection algorithm was developed to overcome these challenges.
Why is CNV detection important when analyzing next-generation sequencing data?
Next-generation sequencing (NGS) is a high-throughput technique that generates high-resolution genomic data which allows for simultaneous detection of many genomic variants, such as SNVs, Indels, and CNVs. CNVs are a structural variation in which DNA segments of one kilobase or larger are present at a variable copy number (duplications or deletions) compared to a reference genome. They have clinical and diagnostic relevance as they have been associated with cancers and rare genetic disorders. Although microarray (or SNP-array) comparative genomic hybridization (aCGH) and multiplex ligation-dependent probe amplification (MLPA) are the gold standards for CNV detection, neither can detect small variations such as SNVs and Indels. The decreasing cost of NGS and the ability to simultaneously detect multiple genomic alterations in a single run have encouraged the widespread use of NGS for CNV detection.
Why are CNVs generally difficult to detect using NGS?
CNVs are challenging to detect via targeted capture because the relationship between sequencing depth and copy number is affected by many sources of bias, e.g., GC content and target region length, capture efficiency, amplification efficiency, DNA concentration, hybridization temperature, nature of capture, batch effects, and so on. These biases result in coverage heterogeneity, even for diploid regions (copy number of 2) and must be accounted for to accurately infer copy number from coverage data.
What challenges are associated with CNV detection in exome data?
On top of overcoming the biases mentioned above, when analyzing the human exome we have the cumulative challenge of sequencing only the protein-coding regions (exons). This results in sparse coverage, as the targeted regions only cover about 1% of the whole genome. Lack of coverage across the entire genomic profile causes us to miss most breakpoints, leaving read depth as the only available information source for CNV detection. Other challenges with detecting CNVs in exome data include the presence of many polymorphic regions for which the normal copy number is already higher or lower than two, and the presence of homologous regions, which is problematic for short read alignment.
How are CNVs detected using the SOPHiA DDM™ Platform?
CNV analysis by SOPHiA DDM™ Platform is performed based on coverage analysis of targeted regions. MUSKAT™ automatically selects reference samples among the samples within the same run to perform normalization. We apply a double normalization to account for both sample-specific and region-specific biases. CNV detection is performed by using a hidden-Markov-model algorithm to find CNVs spanning adjacent regions. Additionally, the algorithm provides quality measures for each sample based on the residual noise.
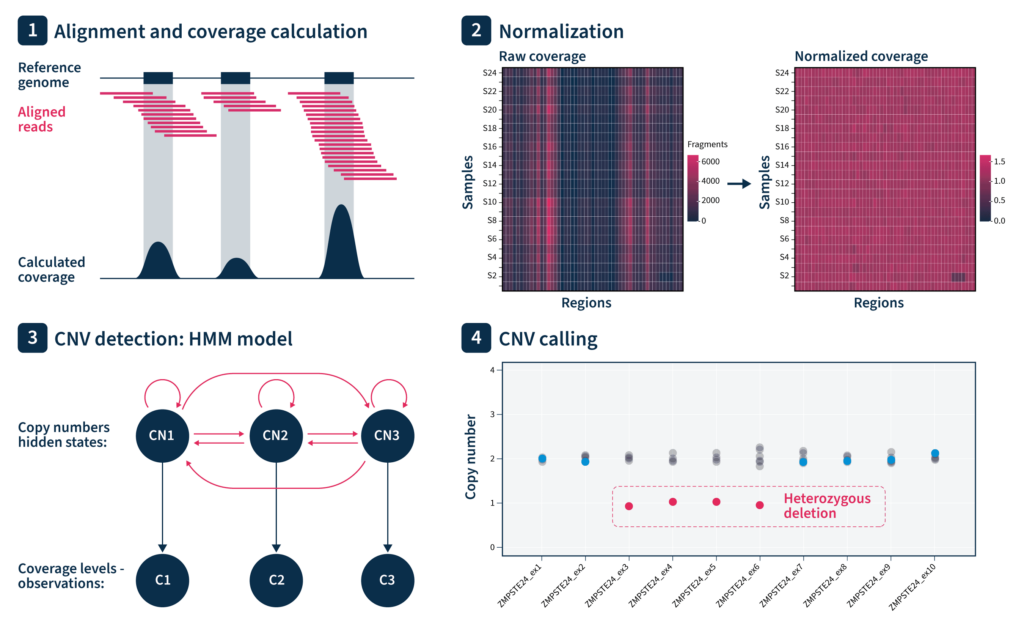
What is the reasoning behind SOPHiA GENETICS’ approach?
Our normalization approach corrects for read-depth variations among regions by leveraging information from different samples in the same run. Assuming that all samples are processed in parallel, the double-normalization step corrects for all sources of targeted sequencing bias mentioned earlier. We also use our knowledge of the genome to curate target regions for each specific exome panel so that regions that would be problematic for MUSKAT™ are excluded, e.g., regions with systematically low coverage, high noise, or polymorphic or homologous regions.
What parameters does the exome sequencing panel need to achieve for good quality results?
Datasets with high coverage and low capture bias achieve high-quality results.
What resolution of CNVs can be achieved?
It depends on the exome panel, but with high-quality panels (good probe design) and deep sequencing depth (~600x), we can achieve even single-exon resolution.
What sets SOPHiA GENETICS’ CNV-calling algorithm apart from others?
Four key features set the SOPHiA GENETICS CNV-calling algorithm apart from others. MUSKAT™:
These four features ensure that we achieve good sensitivity and precision in CNV calling with the SOPHiA DDM™ Platform for Rare and Inherited Diseases, including inherited forms of cancer.
At ACMG 2022, leading geneticists gathered in Nashville, Tennessee and online to share exciting advances in the fields of medical genetics and genomics. The research topics spanned from laboratory research, to prenatal genetics, clinical genetics and therapeutics, public health, and beyond. Here, we summarize hot-off-the-press findings from human exome sequencing, showcasing the potential utility of this technology in the future of healthcare.
Yan Lyu, MD, Peking Union Medical College Hospital, Yulin Jiang, MD, Qingwei Qi, MD, Xiya Zhou, MD, Qi Guo, MD, Na Hao, BS, Juntao Liu, MD
With the increased utility of exome sequencing for prenatal diagnosis, Peking Union Medical College Hospital conducted a large, multi-center prospective clinical study. Lyu and colleagues investigated the diagnostic yield of exome sequencing (via trio analysis) for 306 fetal ultrasound anomalies which were undiagnosed with karyotyping or chromosomal microarray analysis (CMA).
They found that the diagnostic yield of exome sequencing was 39%, and was slightly higher in fetuses with recurrent anomalies (55%) than in those with multisystem anomalies (34%). Among fetuses with isolated anomalies, the diagnostic yield was highest for skeletal (49%) and lowest for cardiac (18%) anomalies. 44% of diagnosed fetuses had a de novo variant and 56% had an inherited variant, with causative copy number variations (CNVs) found in 3.3%. De novo variants were most frequent in fetuses with cerebral (86%), multisystem (71%), or skeletal (65%) anomalies. Causative variants in some genes were present in multiple fetuses – COL1A1 and FGFR3 for skeletal anomalies, TSC2 for cardiac anomalies, L1CAM for cerebral anomalies, and KMT2D for multisystem anomalies (Kabuki syndrome).
These findings demonstrate that exome sequencing can successfully identify the underlying genetic cause of ultrasound anomalies, even when karyotyping or CMA cannot. Furthermore, the details relating to specific anomaly types and causative genes will guide clinicians and parents to manage current pregnancies and inform future pregnancies, while also paving the way for more research in this important field.
Charlotte Sherlaw-Sturrock, West Midlands Genetics Services, Birmingham Women's and Children's Hospital, Birmingham, UK, Helen McDermott, MB ChB, MRCPCH, Julia Baptista, BSc (Hons), PhD, DipRCPath, Lorraine Hartles-Spencer, BSc, Swati Naik, MBBS, DCH, MRCPCH, CSER
National rapid exome sequencing was introduced to NHS England in October 2019 for acutely unwell children with a likely monogenic disorder, or for pregnancies in mothers with a previously affected fetus or child.
The West Midlands Regional Genetics Centre assessed the impact of this rapid exome sequencing service on children and their families. Over the 12-month study period, 95 probands had rapid exome sequencing, 89% with trio analysis. The median turnaround time for preliminary reports was 11 days, with a diagnostic yield of 40% (microarray assisted the diagnosis in 4 patients). The diagnostic rate was highest for patients with neuro-regression, skeletal dysplasia, and neuromuscular or neurometabolic conditions.
The study found that a rapid genetic diagnosis for critically unwell children can have a significant impact on acute and long-term management. A genetic diagnosis helped families to come to terms with their child’s medical condition, enabled referral to the most appropriate specialist, informed clinical management (such as initiation of targeted treatments or transplant), and in some cases helped to predict long-term prognosis while avoiding invasive investigations such as muscle biopsy and post-mortem examination. In addition, recurrence risk counselling was provided, and prenatal diagnosis was made possible for many families.
Overall, NHS England’s rapid exome sequencing service successfully revolutionized the clinical management of many critically unwell children, and will continue to do so.
Wei-Liang Chen, Seattle Children's Hospital and University of Washington, Andrew Timms, PhD, Emily Pao, MPH, Jessica Chong, PhD, Michael J. Bamshad, MD, Debbie Nickerson, PhD, Dan Doherty, MD, Edward Novotny, MD, Russell Saneto, MD, Richard Ellenbogen, MD, Jason Hauptman, MD, Jeff Ojemann, MD, William Dobyns, MD, Kimberly Aldinger, PhD, Ghayda M. Mirzaa, MD
Brain malformations are associated with high rates of morbidity and mortality and their genetic landscape is poorly understood. It is difficult to obtain a molecular genetic diagnosis for individuals with these developmental brain disorders, due to complex clinical and neuroimaging features and their rarity as individual disorders.
To better understand the genetic basis of brain malformations, Seattle Children’s Hospital and the University of Washington conducted a comprehensive analysis of exome sequencing and neuroimaging data. The group enrolled 566 individuals from 526 families for exome sequencing, 78% of which underwent trio analysis. The overall diagnostic yield was 36.7%, increasing to 49.4% when combined with families for whom a candidate gene was already identified. The diagnostic yield was highest (close to 50%) in individuals with megalencephaly and microcephaly, and lowest in those with developmental encephalopathies (25.6%). Pathogenic variants that likely contributed to the onset of certain developmental brain disorders were identified in the novel candidate genes MACF1, CEP85L, RELN, PIK3CA, AKT3, PIK3R2, PDHA1, GRIN2B, TUBB1, and AP1S2.
This detailed analysis of clinical, neuroimaging, and genetics data found that exome sequencing has a high diagnostic yield in individuals with brain malformations and further expands the known genetic and phenotypic spectrum of developmental brain disorders. Furthermore, the results highlight the combinatorial strength of genetic analysis coupled with detailed phenotyping by neuroimaging when diagnosing brain malformations.
Every year, patients, caregivers, clinicians, and researchers celebrate Rare Disease Day on February 28th. Our team at SOPHiA GENETICS continues to support the management of rare and inherited diseases by advancing data-driven medicine.
The winner of this year’s Rare Disease Day quiz is Maria Teresa Bochicchio at IRCCS - Istituto Romagnolo per lo Studio dei Tumori "Dino Amadori" – IRST, Meldola, Italy!
Maria Teresa chose to donate the $500 prize to Genetic Alliance UK.
Thank you Maria Teresa, and thank you to everyone who took part in Rare Disease Day 2022.
The advent of whole-genome sequencing (WGS) ushered in a whole new world for biological research. Since the Human Genome Project was (partially) completed 20 years ago, the cost of WGS has plummeted, now sitting at $1000 or less (vs more than $1,000,000 at its height). However, sequencing a whole human genome is data-heavy and can take 100GB to upwards of 1TB of hard drive space for a single sequencing run, generating a vast amount of data to analyze. Enter the exome.
Whole exome sequencing (WES) sequences all the protein-coding regions (exons) of the genome but avoids the non-coding introns. Coming into vogue in the 2010s, the technique has increased exponentially in popularity in the past decade.1 There are several advantages to WES over other technologies. The exome is only about 1.5-3% of the genome, so focusing on the exome rather than the genome substantially reduces the size and cost of a sequencing run. WES runs come in at a scant few gigabytes of hard drive space, and while the cost is not 1.5-3% of WGS, WES generally costs around a quarter of a full genome.2 Exome sequencing is highly efficient, with diagnostic yields ranging from 20-40% in the clinic,3 compared with yields in the single digits for comparative genomic hybridization arrays (aCGH). Though specific figures vary from study to study, exome sequencing tends to beat microarray-based sequencing methods and targeted next-generation sequencing (NGS) gene panels by a factor of 3-4x in terms of diagnostic yield.4 When using NGS techniques to find pathogenic variants, exome sequencing shines, since 89% of variants reported as pathogenic in NCBI’s ClinVar come from protein-coding regions (at least in Mendelian disorders). That number skyrockets to 99% when neighboring regions are included.5
Starting with exome sequencing can shorten diagnostic time and increase diagnostic yield.
In a diagnostic situation, exome sequencing offers many advantages over targeted panels. A targeted panel is often selected from a phenotype-first point-of-view, where a patient’s clinical presentation guides the selection of a specific set of genes to examine. This approach can, however, be unsuccessful, resulting in either needing a whole new panel or reflexing to exome sequencing. Exome sequencing is the preferred method of diagnosis in several common scenarios: when patients have generalizable symptoms that could have arisen from a variety of underlying conditions; when a disease is genetically heterogeneous or has substantial phenotypic variability (like hydrops fatalis); or if a rare disease has a genetic cause that simply lies outside of the known genes included in targeted panels.6,7 What’s more, starting with exome sequencing can speed up diagnostic time, increasing diagnostic yield by more than 40% in one clinic’s hands. Shortening the time from presentation to diagnosis can radically reduce clinical costs and improve patient outcomes, by quickly pinpointing treatment and care options.
Peculiarities in sequence features can be a problem for any kind of exome sequencing analysis. Both disproportionately high GC or TA content can decrease the accuracy of exome sequencing.8 Why this should be the case is not always particularly clear, though blame commonly falls on PCR or polymerase-based issues (due to the higher melting temperature for GC-rich regions for instance). Careful probe design can help avoid this problem. It can also help attenuate the challenge of capturing sequences located within highly ordered, difficult structures — particularly approaches that use overlapping probes rather than end-to-end or even gapped probes.9
Copy number variations (CNVs), mutations that duplicate or delete segments of DNA, arise in a variety of diseases. Until recently, WES was not the preferred method for CNV detection, since chromosomal microarrays performed better (with a yield of around 20% for chromosomal aberrations reported in 2010)10. WES datasets have, in the past, lacked the consistency between methods, and the high-quality reference material needed for some clinical applications. However, recent work proposed changing the order of workflow and incorporating sequencing data from outside WES databases,11 making WES a powerful technique for detecting pathogenic CNVs.
For a wide variety of rare diseases such as metabolic, neurological, and developmental disorders, the precise causative genetic mutation has often only ever been seen once – 69.6% of cases in Orphanet (a rare disease database) have only one documentation.12 Many rare diseases can arise from a variety of contributing genes along a signaling pathway. In these situations, past efforts like those using traditional Sanger sequencing can often fail to pick up a causative variant, but there is a litany of examples of exome sequencing succeeding in their stead.13,14
Although exome data requires heavy processing before analysis, the researcher does not necessarily require a bioinformatics skill set. Much of the processing, like trimming low-quality bases at the end of reads or detecting and deleting adapters, can be achieved automatically using specialized analytic software.
Determining which variants are pathogenic and which are not can be a difficult task. The drawback, of course, of WES versus a targeted gene panel, is that there are much more data to deal with. WES runs commonly find tens of thousands of variants - in extreme cases, hundreds of thousands.15,16 This can introduce an order of magnitude of noise into a dataset. Applying a few key rules and variant filtering strategies can reduce the number of variants by 90-95% so that researchers might then be faced with 150-500 candidate mutations, a much more manageable number.16 Further investigation requires consulting scientific databases and repositories to determine whether the detected exonic variants are pathogenic, benign, or variants of unknown significance (VUS). Unfortunately, there is no single, all-encompassing database that contains all the information needed to interpret variants. That means, unless researchers have access to software that taps into tens of different databases, it will take substantial time to manually check through one by one. In addition, despite the vast amount of information available, a significant proportion of detected variants are still VUS, highlighting the ever-growing need for more research in the field.
The SOPHiA DDM™ Platform for rare and inherited disorders accurately detects a range of variant classes, with high sequence coverage uniformity even in complex and GC-rich regions. Where multiple combinations of NGS technologies can introduce artifacts and inconsistencies, the SOPHiA DDM™ Platform filters the noise and bias to deliver advanced analytical performance independent of the input. This high-quality, noise-filtered output is used to accurately detect CNVs with exon-level resolution. The platform even covers ∼200 variants in non-coding regions and the entire mitochondrial genome to ensure comprehensive exome analysis. No matter the variant type — SNV or CNV — SOPHiA GENETICS’ analytics provide optimized variant detection in a single experiment.
Where pinpointing pathogenic mutations can be difficult, SOPHiA DDM™ complemented by Alamut™ Visual Plus can help cut through the noise and enable a deep exploration of variants. Together, the two technologies annotate variants with information from more than 55 world-renowned biological databases and repositories, including missense and splicing predictors. The SOPHiA DDM™ Platform offers additional filtering features such as Virtual Panels to limit interpretation to genes associated with specific disorders, Cascading Filters to reduce analysis to variants with specific characteristics, and Familial Variant Analysis for consideration of parental samples and inheritance mode. For on-the-ground level accuracy, users benefit from becoming members of the SOPHiA GENETICS Community, where experts flag variant pathogenicity to improve interpretation, even of VUS. Finally, for efficient and user-friendly interpretation, Alamut™ Visual Plus enhances the visualization of variants in a comprehensive full genome browser.

Innovations in sequencing and analysis aren’t slowing, and researchers need tools to keep afloat in the flood of data. Analytical technologies like SOPHiA DDM™ and Alamut™ Visual Plus are ideal parts of a researcher’s arsenal to find pathogenic variants and get clear answers from complex WES datasets.
Within every cell in the human body, there are thousands of mitochondria. They are, for all intents and purposes, little proto-bacteria subsumed into your cells, providing them with energy in an ancient symbiotic relationship — the cell sheltering the mitochondria, and the mitochondria producing energy for the cell by performing aerobic respiration. Each mitochondrion has its own tiny circular genomes , which means that instead of just a solitary genome in the nucleus, each eukaryotic cell is studded with thousands of copies of micro-genomes (creating “heteroplasmy”, where one cell can contain multiple non-identical genomes). One of the earliest descriptions of mitochondria came in 1890, when Richard Altmann called them “bioblasts” or “life germs”.1,2 The description of the first mitochondria-based disease came 70 years later in 1962, based partly on analysis of a hyperthyroid patient whose tissue displayed physically abnormal mitochondria and had biochemically “loose coupling” of aerobic respiration.3
The early landmark work on mitochondrial genomes started arriving around the late 1970s and early 1980s. The mitochondrial genome was in fact the first complete eukaryotic genome ever sequenced. This was in a pre-PCR, Sanger-sequencing world when the idea of even thermal cycling hadn’t taken hold. The first paper that described the mitochondrial genome’s unusual properties is an interesting time capsule for how laborious molecular biology and genome sequencing was 40 years ago (this is actually a separate paper from the one describing the complete mitochondrial genome, which came a few years later in 1981).4,5 Upwards of 80 proteins participate in oxidative phosphorylation (one of the component steps of aerobic respiration), with the mitochondrial genome encoding 13 of them.6 Though the exact percentage of mitochondria inherited from either parent is debatable, the vast, vast majority is inherited from the mother, and so maternal mitochondrial genetics will dominate the child’s mitochondrial genetics.7
Mutations in the mitochondrial genome cause a litany of human diseases, affecting 1 in 5000 people.8,9 Mitochondrial disorders are particularly common, as the mitochondrial genome experiences mutations at a far higher rate (commonly thought to be between 10- and 100-fold higher) than the nuclear genome.10 This susceptibility to mutation is thought to arise from mitochondrial DNA’s (mtDNA’s) proximity to radical oxygen species produced during respiration. The symptoms of mitochondrial disorders are diverse, from neurological disorders like migraines, strokes, epilepsy, and dementia; damage to the senses; to essentially any organ you can think of, resulting in diseases as diverse as diabetes and thyroid disease, renal failure, heart failure, and disorders of speech. There have been demonstrated instances of patients with complex metabolic disorders, whose genetic etiology was left unexplained by nuclear sequencing and was only clarified when their samples were re-sequenced to examine their mitochondrial genomes.
Inherited mitochondrial disorders can occur in any tissue type. A small load of inherited mtDNA mutations tends not to create medical conditions in a person’s lifetime, though determining what the threshold is for inherited mutations that do cause disease is difficult to pin down, and likely varies by the tissue that a mutation occurs in. Mitochondrial disorders are not only inherited but can arise due to a previously unseen mutation, which can lead to a complicated diagnosis.
Mutations in the mitochondrial genome cause a litany of human diseases, affecting 1 in 5000 people
In the era before robotics, Sanger sequencing was a laborious task. The mitochondrial genome was cut into pieces using specific restriction enzymes, cloned into a plasmid, and sequenced using radioactively labeled primers. 16,569 base pairs were read and recorded through Sanger runs on slab gels,5 which are as thin and fragile as tissue paper and can accommodate 100-200 bases per run at the most. It was brute-force methods that both sequenced the entire mitochondrial genome and found its unusual properties, like its reassignment of the UGA stop codon to tryptophan and its unique suite of tRNA genes that don’t match the nuclear genome.
In the era before laboratory computers, sequence alignment, and genome browsers, mutations in mtDNA were detected by “sequence gazing”: just looking at sequences until you spotted differences. A missense mutation had to be found by hand, and a frameshift analyzed by eye and compared against the unique codon table of the mitochondria. The diagnostic yield for mitochondrial disorders in adults can be as low as 11% using Sanger sequencing, but as high as 40-60% using next generation sequencing (NGS).11
Thankfully, the introduction of NGS has made both the lab work and analysis of mitochondrial genetics data much, much more approachable, no matter the size and background of the lab. NGS techniques make detecting uncommon mutations in large pools of data — like, say, a large and heterogeneous collection of mitochondrial genomes — a magnitude of order easier than Sanger sequencing, on top of being able to multiplex a variety of different samples in a single sequencing run. Rare, disease-causing mitochondrial genome mutations may be below the limit of detection of traditional Sanger sequencing, in some cases requiring that NGS be performed.
In human cells, there is only one nuclear genome, containing about 3 billion bases. The mitochondrial genome is only 16,659 base pairs long, but the varied number of mitochondrial genomes per cell, and the variation between each mitochondrion, makes the sequencing depth and coverage uniformity made possible by NGS critical. The relatively higher number of mitochondrial (versus nuclear) genome copies also contributes to increased mitochondrial DNA mutational rates — more copies, more chances for error. Depending on the sample type, there is huge variability in the number of mitochondria and thus the copy number of mitochondrial genomes. Less energy-intensive tissues like the lungs can have as few as several hundred mitochondria per cell, whereas tissues that require huge amounts of energy, like the liver, heart, and skeletal muscle, can have 1000-7000 mitochondria per cell.12 Furthermore, the number of mitochondria per cell can itself be a marker for disease (called mitochondrial DNA depletion syndromes), though this can be detected using quantitative PCR, without the need for a sequencing run.
Heteroplasmy, the variation in abundance of pathogenic variants due to the fact that there are thousands of distinct mitochondrial genomes per cell, can make analysis difficult. On top of that, nuclear mitochondrial sequences (NUMTs), translocations of mitochondrial DNA to the nucleus, can make capturing data specific to the mitochondrial genome more complex than simple whole-cell sequencing. Finally, even if a genetic test is ordered in the clinic, these normally do not include the mitochondrial genome, thus completely discounting mitochondrial disorders from the outset.

At SOPHiA GENETICS, we offer integrated mitochondrial testing as part of our clinical exome and whole exome solutions, enabling the identification of pathogenic variants in the human exome and mitochondrial genome in a single experiment. Our panel design and the sophisticated algorithms used by the SOPHiA DDM™ platform address the unique challenges associated with the mitochondrial genome, such as the high and variable amount of mtDNA and heteroplasmy, to provide coverage uniformity and accurately and confidently identify variants in mtDNA in a streamlined analytical workflow.
Software like Alamut™ Visual Plus can take the eyestrain out of sequence gazing by allowing visualization of detected variants in a comprehensive full genome browser. SOPHiA DDM™ and Alamut™ Visual Plus can simplify the complex and time-consuming assessment of mitochondrial variant pathogenicity by pre-classifying and annotating variants with curated, up-to-date information from literature sources such as PubMed® and MasterMind®, and genomic databases such as ClinVar, OMIM®, and dbSNP, in a single, user-friendly environment.
Where sequencing and gazing at a mitochondrial DNA sequence in the 1980s could have taken weeks of squinting at gels, the SOPHiA DDM™ platform turns around sequencing data in a matter of hours, and Alamut™ Visual Plus can accelerate variant interpretation to a few clicks.
Last week, more than 7000 people gathered from around the globe to digitally attend the Festival of Genomics & Biodata (FoG). The diverse sessions spanned the entire genomics workflow, with the ultimate mission of sharing knowledge to benefit patients. Here, we summarize three FoG talks on the future of genomics for rare and inherited disorders: we aim to show the big picture, walk you through a specific medical condition, and then provide an in-depth description of an individual case.
Over the last 30+ years, there has been an almost exponential increase in the number of rare and inherited diseases with a molecular diagnosis. Between 1989 and today, ∼7000 new gene-phenotype descriptions and 600 targeted therapies have been identified, with orphan drugs representing >50% of all U.S. drug approvals. Encouragingly, the life expectancies of people with these conditions are also steadily increasing, largely due to healthcare professionals collaborating on developing standards of care for similar conditions.
Rare and inherited diseases require a unique model of science and medicine due to a set of unique challenges. Due to their genetic nature, most are not limited to specific organs or systems and span an entire lifetime. In U.S. children’s hospitals, 34% of patients have inherited disorders which account for 81% of hospital bills, a clear financial impact on the health system. Rare disease research is based on small numbers of patients, and the care model is complex, involving multiple specialties with workforce challenges, so that patients and their families are often the experts on their condition.
Rare disease research is based on small numbers of patients, and the care model is complex, involving multiple specialties with workforce challenges, so that patients and their families are often the experts on their condition.
Collaboration, new care models, and technology are needed to navigate these unique challenges associated with rare and inherited diseases. New tactics to speed up and improve the patient pathway include digital triage, newborn screening, facial recognition software for identification of specific syndromes, more research to identify gene-phenotype relationships for molecular genetic testing, and increased involvement of primary care providers throughout the pathway. Telemedicine (fully trialled during the COVID-19 pandemic) also has a multitude of benefits, such as decreases in wait times, increased convenience, the ability for patients with autism to be seen in the comfort of their own home, and the ability for multiple specialists to easily collaborate remotely. In addition, certain barriers to care are removed, such as transportation (especially for geographically remote patients), childcare, missed work, and infection risk for vulnerable patients.
Continued innovation and collaboration provides optimism for a brighter future for patients with rare and inherited diseases.
A new preventive genomics cardiology clinic at Mass General empowered people to better understand, predict, and prevent heart attacks based on their genetic information.
Most people are aware of the clinical and lifestyle factors associated with heart attacks – for example, smoking, diet, exercise, and cholesterol levels. Genomic factors such as monogenic and polygenic drivers also play a critical role.
Monogenic risk of heart attack results from rare but high-risk single gene defects in the LDL “bad” cholesterol pathway. Whereas polygenic risk results from the cumulative effect of millions of relatively common, moderate-risk variants. Polygenic risk scoring identifies people who are at risk of having a heart attack but could have otherwise “flown under the radar” as their LDL cholesterol levels were normal. Indeed, out of every 100 people who have a heart attack at a young age, 2 have monogenic and 20 have polygenic genomic drivers.
Out of every 100 people who have a heart attack at a young age, 2 have monogenic and 20 have polygenic genomic drivers.
Integrating these genomic drivers with clinical and lifestyle risk factors is the most accurate way to identify people at high risk of having a heart attack. Indeed, a favorable lifestyle halves the risk of coronary events in people with a high monogenic or polygenic risk.
At the preventive genomics clinic at Mass General, 45 people took a new genetic test for heart attack risk. About half were referred by their cardiologist and half self-referred. 25% of all participants were found to have a high genomic risk for heart attack: 20% due to polygenic drivers and 5% due to combined monogenic and polygenic factors. Clinical action was taken for half of these high-risk participants – e.g., some were given statins for the first time, had a statin dose increase, or had coronary imaging.
These changes in clinical care based on a simple genetic test could ultimately prevent heart attacks in people who did not previously know that they were at risk.
Peter’s wife and son have undiagnosed, debilitating, rare and inherited conditions, and Peter is determined to do everything in his power to provide them with the care that they desperately need. In fact, this devoted husband and father spent 8 hours a day for the past 6 months pouring over sequencing data to try and find the cause of their suffering.
Now in their 70’s and 30’s, Peter’s wife and son have been chronically ill since puberty. Hormonal imbalance, urticaria, clinical depression, chronic pain, exercise intolerance, extreme fatigue, edema, and myotonia are just a few of their symptoms. All of which are exacerbated by the stress of being in constant pain and discomfort, and vary from one day to the next. Over the years, a complex combination of drugs (muscle relaxants, antihistamines, opioids, beta-blockers…) has been cobbled together to help ease some of their symptoms, but not completely or consistently.
The family’s diagnostic odyssey is confounded by the public health system, which even with the very best intentions, makes it extremely difficult for patients with intermittent symptoms that span numerous specialties to get the help that they need. Neurologists, psychiatrists, pain specialists, and pulmonologists have all contributed to the pair’s growing library of medical records, but long waiting times and a lack of the “full picture” severely limits the quality of care that they can provide.
In a recent attempt to end the diagnostic odyssey, the family had their whole genomes sequenced and analyzed, but to no avail. This is when Peter decided to take matters into his own hands. He sat down with the raw sequencing data and started to analyze it himself. With just a basic background in science and bioinformatics, he took it upon himself to examine their exome sequences line by line to try and identify pathogenic variants - using various tools and consulting various experts along the way. Truly an inspiration, Peter has started to identify variants that look to be involved in causing his family’s rare disease but has a long way to go. He hopes that the Festival of Genomics & Biodata will connect him with more researchers and experts that can help with his mission. Peter has hope in the power of genomics for people with rare and inherited diseases – he will not give up.
Peter has hope in the power of genomics for people with rare and inherited diseases – he will not give up.
SOPHiA GENETICS products are for Research Use Only and not for use in diagnostic procedures unless specified otherwise.
SOPHiA DDM™ Dx Hereditary Cancer Solution, SOPHiA DDM™ Dx RNAtarget Oncology Solution and SOPHiA DDM™ Dx Homologous Recombination Deficiency Solution are available as CE-IVD products for In Vitro Diagnostic Use in the European Economic Area (EEA), the United Kingdom and Switzerland. SOPHiA DDM™ Dx Myeloid Solution and SOPHiA DDM™ Dx Solid Tumor Solution are available as CE-IVD products for In Vitro Diagnostic Use in the EEA, the United Kingdom, Switzerland, and Israel. Information about products that may or may not be available in different countries and if applicable, may or may not have received approval or market clearance by a governmental regulatory body for different indications for use. Please contact us to obtain the appropriate product information for your country of residence.
All third-party trademarks listed by SOPHiA GENETICS remain the property of their respective owners. Unless specifically identified as such, SOPHiA GENETICS’ use of third-party trademarks does not indicate any relationship, sponsorship, or endorsement between SOPHiA GENETICS and the owners of these trademarks. Any references by SOPHiA GENETICS to third-party trademarks is to identify the corresponding third-party goods and/or services and shall be considered nominative fair use under the trademark law.