Menu
Menu
Tell us a bit about your role.
I am a Senior Clinical Genomic Variant Analyst at the Broad Clinical Labs – a CLIA/CAP certified lab at Broad Institute of MIT and Harvard. Broad Clinical Labs offers fee-for-service clinical genomic testing, and I am part of the clinical interpretation team for whole genome sequencing. Clinical testing can be ordered from our laboratory by clinicians; our lab performs whole genome sequencing and provides technical genome data and an interpretative report. Our team’s goal is to review the genome data and find reportable variant(s) to share with clinicians.
What type of cases do you work with?
Some of the testing we perform is panel based, but mostly we work with whole genome sequencing data. Because we do not bill insurance, we create relationships with individual institutions or organizations and serve as their sequencing and interpretation partner. Currently, some of the projects we are partnering on include cases focused on rare disease, a CICU cohort, and neurodevelopmental disorders. We use Alamut™ Visual Plus as an add-on tool to our existing variant interpretation workflow when we need additional information about an identified variant.
What are the biggest benefits of Alamut™ Visual Plus that help you overcome your variant interpretation challenges?
Seeing and understanding the genomic context that a variant exists within can be critical for interpretation. Alamut™ Visual Plus makes it easy to see the context at a glance, providing link-outs to other databases for even more detailed information.
Alamut™ Visual Plus is especially helpful when I’m trying to assess the potential impact of splicing. Alamut™ Visual Plus provides calculated splice predictor scores, but I also want to see exactly where in the gene the variant falls in relation to the exon and to visualize the potential impact of exon skipping and whether it’s likely to result in an in-frame or out-of-frame change. Alamut™ Visual Plus makes this easy to visualize.
Can you share an example where this made a difference?
I was recently assessing a splice variant in a gene known to cause a disease that fit with the given phenotype. The variant was +3bp away from a codon, but this alone wasn’t sufficient to make it reportable. After visualizing the splicing impact of the variant within Alamut™ Visual Plus to confirm the potential for out-of-frame exon skipping, I felt more confident to report the variant as a VUS of interest.
Do you have another example where Alamut™ Visual Plus helped you assess a variant?
In another recent case, I had found what looked to be a potentially relevant variant. This variant had been reported using different historical nomenclature in several publications. I came across one paper with a series of cases and variants, that included a variant at both the historic nomenclature and the current nomenclature position. Alamut™ Visual Plus let me easily view and scan the context of the amino acid sequence across different transcripts to appropriately anchor myself to the right reference. This allowed me to confirm I was using correct variant information from this publication in my interpretation.
We’d like to thank Katherine Lafferty for her time and for sharing her experience. We look forward to continuing our conversations with Broad Clinical Labs! Click here to learn more about Alamut™ Visual Plus and request a free trial.
Assessment of Homologous Recombination Deficiency (HRD) and BRCA mutational status in high-grade serous ovarian carcinoma (HGSOC) samples has become crucial for personalized medicine, guiding treatment decisions, and predicting response to specific therapies, such as PARP inhibitors.
The aim of this multicentric study, published at this year’s European Society of Gynaecological Oncology (ESGO) conference was to assess the reliability and reproducibility of SOPHIA DDM™ Dx HRD Solution across three different hospitals (Hospital del Mar, Hospital Vall d’Hebrón and Hospital Clinic de Barcelona) to consider its implementation as a decentralized solution in routine molecular diagnostics.
Our Spanish team led by Giovanni Velotta, CS Manager at SOPHiA GENETICS, was happy to meet Dr. Gardenia Vargas, Molecular Geneticist and Responsible for Hereditary Cancer and Rare Disease Molecular Diagnosis at Hospital del Mar in Barcelona, Spain, who joined us for an insightful interview, sharing her experience in implementing SOPHiA DDM™ Dx Homologous Recombination Deficiency Solution.
Before I answer I would like to thank you and thank AstraZeneca for their support in this study. I must mention that all my answers represent my own perspectives and not necessarily the official stance of the hospitals involved in this project.
The study aimed to investigate the feasibility and effectiveness of implementing the SOPHiA DDM™ Dx HRD (Homologous Recombination Deficiency) Solution within the clinical setting of three hospitals. This includes evaluating various aspects such as the practicality of integrating the test into existing diagnostic workflows, the accuracy and reliability of test results, the impact on patient care and outcomes, and the potential benefits and challenges associated with in-house testing. The study seeks to provide valuable insights into the utility and suitability of adopting the SOPHiA DDM™ Dx HRD (Homologous Recombination Deficiency) Solution as a routine diagnostic tool for identifying HRD in ovarian cancer patients.
First of all, I must say that all three hospitals worked equally on this project, and the idea of doing it was born before I was incorporated to it. Beatriz Bellosillo from Hospital del Mar was the principal investigator, and we had different responsibilities such as protocol writing, ethics approval coordination, securing funding to support the study's activities, and management and allocation of some resources like sequencing supplies.
And of course, it was truly a rewarding experience collaborating with renowned hospitals and esteemed colleagues.
Currently, while we have conducted a multicenter study to evaluate the feasibility of implementing the SOPHiA DDM™ Dx HRD Solution, we have not yet implemented it into routine diagnostic practice at my hospital.
The implementation process requires significant financial support, particularly for acquiring the necessary equipment, reagents, and personnel training. Additionally, to ensure cost-efficiency and timely responses to patients, we need to conduct sequencing runs with a minimum number of samples per run.
Unfortunately, our hospital alone does not have sufficient sample volume to meet this requirement. Therefore, we are actively seeking financial support from government agencies to fund the implementation of the study. Additionally, we are exploring collaborative efforts with other hospitals or healthcare institutions to pool together an adequate number of samples for sequencing runs, thus reducing costs per sample and ensuring a rapid response to patients.
By securing both financial support and an adequate sample volume, we aim to overcome these logistical challenges and proceed with the implementation of the SOPHiA DDM™ Dx HRD Solution into our diagnostic routine, ultimately enhancing our ability to provide efficient and effective patient care.
It highlights the importance of collaboration among hospitals, government agencies, and other stakeholders to support the implementation of advanced diagnostic technologies into routine clinical practice.
Overall, the study provides valuable insights into the challenges and opportunities associated with integrating advanced genomic testing into routine diagnostics, with the aim of improving patient care and outcomes.
This collaborative study concludes that SOPHIA DDM™ Dx HRD Solution provides reliable and consistent results across different hospitals and sequencing runs.
These findings contribute to the growing body of evidence supporting the use of the decentralized SOPHiA DDM™ Dx HRD Solution in clinical settings for genomic analysis.
DISCLAIMER: SOPHiA GENETICS products are for Research Use Only and not for use in diagnostic procedures unless specified otherwise. SOPHiA DDM™ Dx Homologous Recombination Deficiency Solution is available as CE-IVD product for In Vitro Diagnostic Use in Europe and Turkey.
Explore this infographic summary to gain insights into the key findings from Pozzorini et al.’s publication on the GIIngerTM deep learning algorithm for prediction of HRD status and patient response to PARPi treatment in ovarian cancer.
Click here to read the full publication.
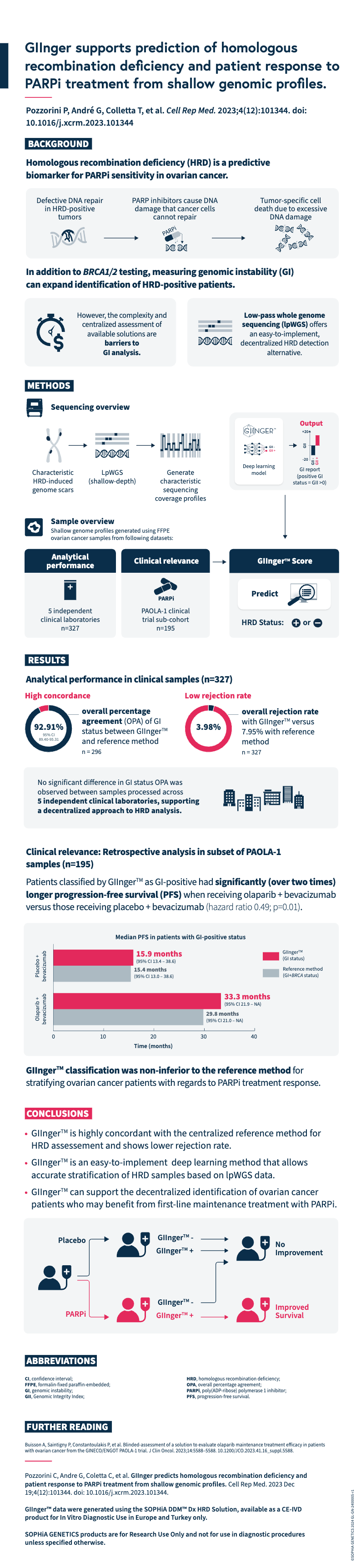
Pozzorini C, Andre G, Coletta C, et al. GIInger predicts homologous recombination deficiency and patient response to PARPi treatment from shallow genomic profiles. Cell Rep Med. 2023 Dec 19;4(12):101344. doi: 10.1016/j.xcrm.2023.101344.
GIInger™ data were generated using the SOPHiA DDM™ Dx HRD Solution, available as a CE-IVD product for In Vitro Diagnostic Use in European Economic Area (EEA), the United Kingdom and Switzerland . SOPHiA GENETICS products are for Research Use Only and not for use in diagnostic procedures unless specified otherwise.
What is liquid biopsy?
Liquid biopsies enable analysis of biofluids, typically blood, to examine biomarkers shed by solid tumors into circulation1. They can detect actionable genomic alterations in a non-invasive way, providing valuable insights to facilitate early cancer detection and disease monitoring2.
Tumor-derived biomarkers that are a source for liquid biopsy analysis include (Fig. 1):
CTCs are initially released from primary tumors in the tissue, travel through the bloodstream, and account for the development of metastatic tumors at distant sites in the body. As they are live cells, they have the potential to be used for functional analysis such as therapy sensitivity/resistance evaluation3. However, CTCs are rare events in the blood, which makes them difficult to identify and characterize in routine clinical practice1.
EVs are membrane-enclosed structures containing proteins, genetic material, and lipids that can provide biological information on the cell of origin4. Due to their role in pathological processes, EVs are an attractive analyte for liquid biopsy, but their isolation and purification is technically challenging1.
cfDNA refers to DNA fragments that are freely circulating in the bloodstream, primarily originating from normal cells5. Circulating tumor DNA (ctDNA) is the small portion of cfDNAthat derives from tumor cells or CTCs undergoing cell death (i.e. apoptosis or necrosis)5. There are well-established methods for isolating cfDNA, and for analyzing it using methods such as PCR and next-generation sequencing (NGS)-based tests6, making it an ideal and feasible substrate for routine genomic analysis.
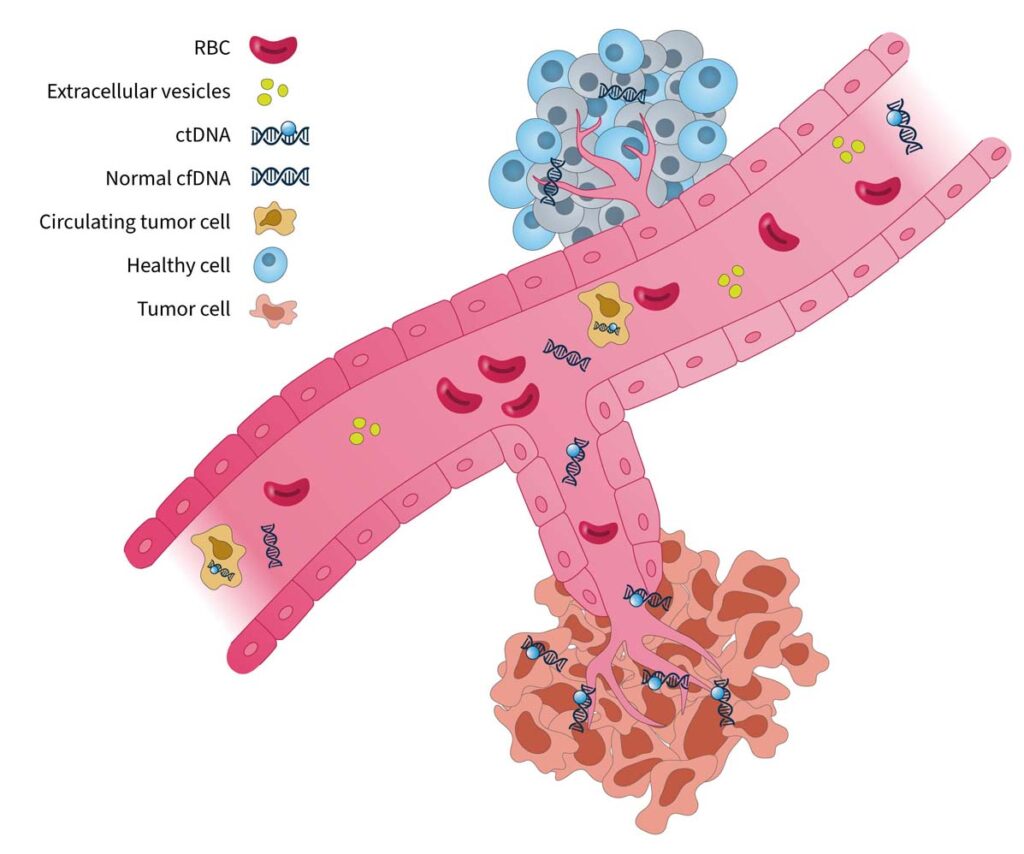
Figure 1. Blood-based cancer biomarkers in liquid biopsy7. RBC, red blood cell.
The analysis of cell-free DNA is a promising method for guiding clinical decisions and can complement current standard-of-care practices8.
What are the clinical applications of liquid biopsy?
In the era of precision medicine, tumor molecular profiling is a critical tool to identify targetable alterations and guide treatment decision-making9. Tissue biopsy is currently the gold standard for tumor profiling8; however, there are limitations associated with this approach:
Liquid biopsy has the potential to be a transformative tool in clinical oncology, showing promise for applications in many stages of cancer management (Fig. 2):

Figure 2. The advantages and clinical utility of liquid biopsy in the cancer care journey10–15.
Innovations in liquid biopsy analysis over the past decade have led to the regulatory approvals of blood-based tests to guide treatment for NSCLC, prostate, breast, and ovarian cancers16. Clinical guidelines have also provided expert recommendations for its use in specific clinical scenarios8,15. Despite great advances in technology and its increasing utility in clinical practice, there are still challenges to overcome when using liquid biopsy to identify clinically relevant information.
Overcoming “fisherman’s luck” in liquid biopsy
One challenging aspect of liquid biopsy analysis is that ctDNA concentration varies greatly across cancer types and between patients17. In patients with cancer, the quantity of ctDNA in the blood can be impacted by several factors, including histology, tumor site, clinical factors (age, sex, treatment history, etc.), and ctDNA fragmentation17. Therefore, it is important to have a robust test to detect clinically relevant variants, even at low ctDNA concentrations against a cfDNA background.
Another factor that may impact liquid biopsy analysis is the presence of clonal hematopoiesis of indeterminate potential (CHIP). In healthy individuals, the majority of cfDNA arises from hematopoietic cells (i.e. stem cells in the bone marrow that give rise to other blood cells)18. Normal hematopoietic cells accumulate somatic mutations during aging, known as CHIP, which are technically indistinguishable to tumor-specific mutations in NGS assays18,19. It is important that the biological noise caused by CHIP signals are removed in liquid biopsy analysis to eliminate false positive variant calls and give an accurate representation of disease burden19,20.
These biological confounders can make “fishing” for clinically relevant information in cfDNA a challenge (Fig. 3). For example, if a patient has high disease burden, there is likely more ctDNA available to analyze, which makes it easier to “catch” what you are looking for. However, if there is less ctDNA and more biological noise, you may need to modify your tools and approach to improve your yield.
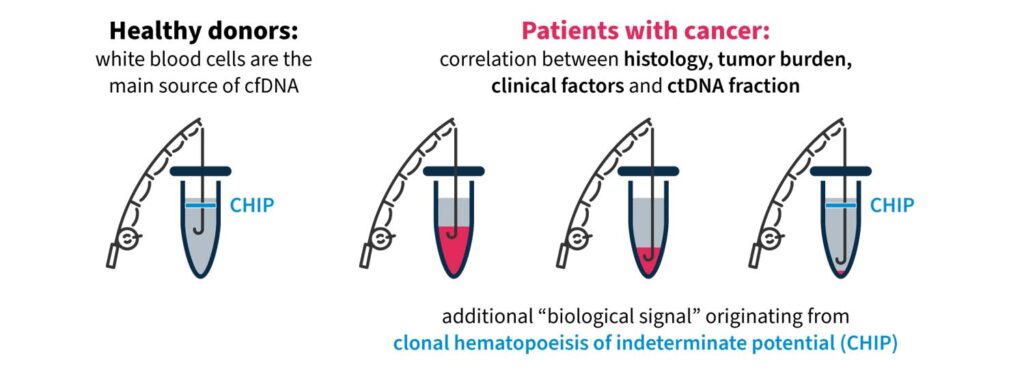
Figure 3. “Fishing” for clinically relevant information in liquid biopsy can be complicated by biological confounders17,18.
Highly precise and sensitive liquid biopsy technologies are needed to overcome “fisherman’s luck” and detect rare, causative variants and disease burden in cfDNA. Guidelines issued by the ESMO Precision Medicine Working Group on the use of cfDNA assays in clinical practice discuss the need for advanced techniques capable of capturing spatial and temporal tumor heterogeneity and reducing rates of false negatives8.
Pioneer innovation with SOPHiA DDM™ for Liquid Biopsy
SOPHiA GENETICS is at the forefront of innovation in liquid biopsy technology for tumor profiling. The advanced proprietary algorithms of the SOPHiA DDMTM Platform empower clinical researchers to reveal deep genomic insights from cell-free DNA samples.
With a streamlined, sample-to-report NGS workflow, you can:
In addition, we are excited to be collaborating with Memorial Sloan Kettering Cancer Center (MSK) to decentralize MSK-ACCESS® for liquid biopsy, designed to provide a maximum coverage of cancer disease variants in ctDNA20. By combining MSK’s clinical expertise in cancer genomics, the predictive algorithms of the SOPHiA DDM™ Platform, and the power of the global SOPHiA GENETICS community, the collaboration aims to expand access to precision cancer analysis capabilities worldwide.
Read more about how you can enhance your analytical capabilities and advance your clinical research here.
SOPHiA DDM™ Community CLL Clonality Solution is an all-in-one application developed and tested by European Hemato-Oncology experts within the SOPHiA GENETICS Community for the analysis and stratification of CLL cases. This sample-to-report approach combines an optimized next-generation sequencing (NGS) workflow and the powerful analytics of the SOPHiA DDM™ Platform.
In this study, we demonstrate the effectiveness of the SOPHiA DDM™ application in generating a comprehensive report on the genomic status of CLL in a fictitious case. Download the application note and learn more about the SOPHiA DDM™ Community Clonality Solution CLL.
What is cascade testing?
Cascade testing is the practice of offering genetic testing to relatives of known carriers of pathogenic variants associated with autosomal dominant conditions. In oncology, cascade testing is performed in families affected by hereditary cancer syndromes1. The most common include hereditary breast and ovarian cancer syndrome (HBOC), Lynch syndrome (LS), familial adenomatous polyposis syndrome, hereditary pancreatic cancer syndrome and gastric cancer syndrome2,3. Testing for variants associated with HBOC and LS belongs to the so-called Tier 1 testing, i.e., genomic applications, the implementation of which is supported by robust evidence4.
Cascade testing starts with first-degree relatives (parents, siblings, children) of index cases (i.e., the family member in whom a pathogenic variant was identified) and then proceeds to second- (grandparents/grandchildren, aunts/uncles, nieces/nephews, half-siblings) and third-degree relatives (great-grandparents/great-grandchildren, first cousins)1.
Most hereditary cancer syndromes follow the autosomal dominant inheritance pattern. Therefore, the first-, second- and third-degree relatives have, respectively, a 50%, 25%, and 12.5% probability of inheriting the predisposition to develop cancer (see Figure 1)1. For some pathogenic variants of genes associated with hereditary cancer syndromes, such as BRCA1/2, the penetrance is high5. Establishing accurate estimates of penetrance and relative risk for genes implicated in hereditary cancer syndromes is an ongoing task3.
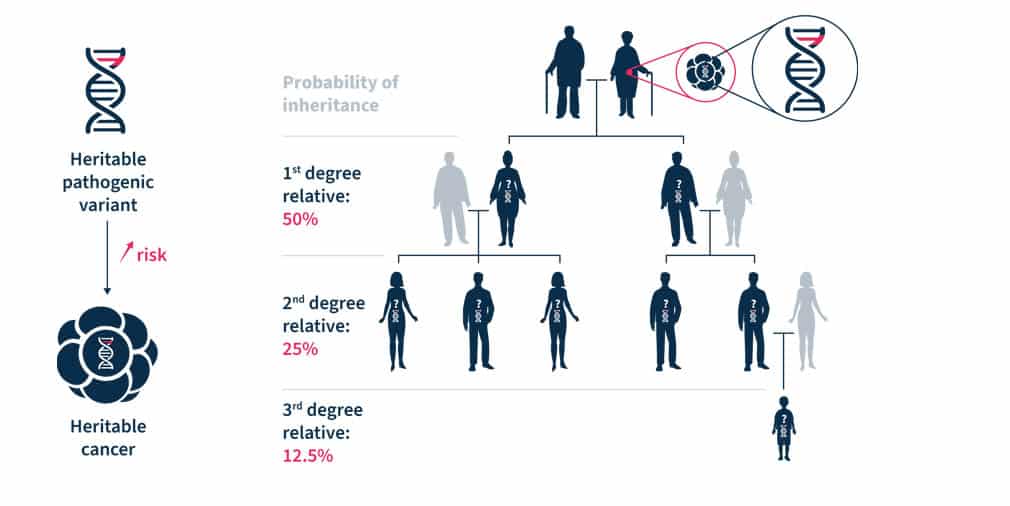
Figure 1. Heritable pathogenic variants increase the risk of developing cancer at a younger age (left). Cascade genetic testing is the practice of testing the relatives of known carriers (right)1.
Why is cascade testing important?
At the level of an individual and their family, cascade testing has two important goals. The first goal is to identify relatives that carry the familial pathogenic variant and require personalized cancer risk management1. The second goal is to exclude the non-carriers from intensive cancer surveillance and prevention interventions1. The detection of pathogenic variants in individuals at a reproductive age may lead to decisions of assisted reproduction or prenatal diagnosis. In the case of actionable monogenic conditions, cascade testing may reduce adverse health outcomes in cohorts of relatives1.
At the societal level, cascade testing has important clinical and research implications for oncology. It can further our knowledge of hereditary cancers and is a cost-effective way of identifying unaffected individuals at-risk, thus, providing important information to plan long-term resources necessary to cope with hereditary cancers. Moreover, today’s testing is needed to tailor future approaches in cascade testing1.
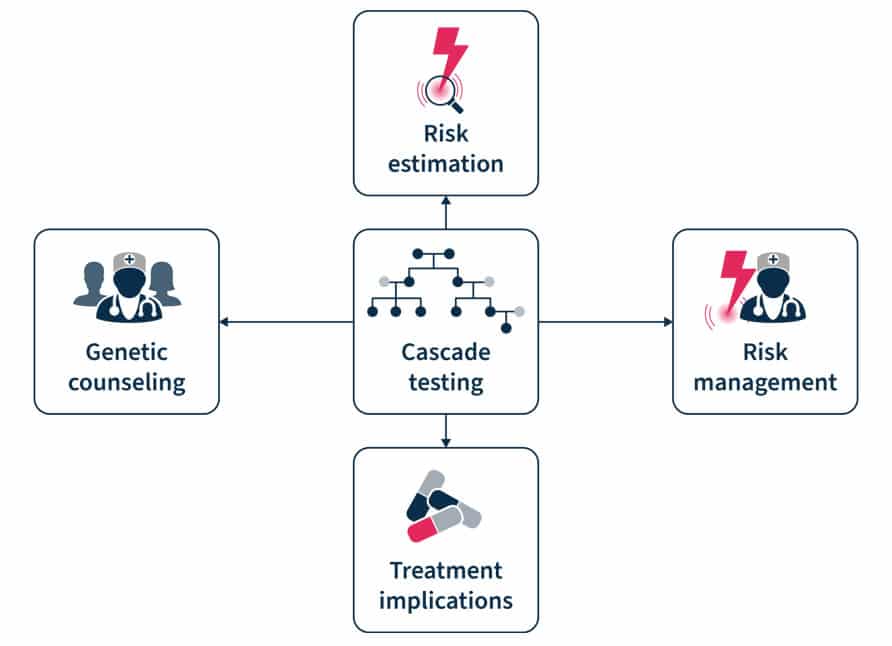
Figure 2. Cascade testing involves genetic counseling before and after the test, risk estimation and management, and has treatment implication7.
What are the barriers to cascade testing?
Despite the advantages of cascade testing, its uptake is low. The reported rates of uptake of cascade testing in HBOC and LS equals ~50% and the underutilization of testing results in missed opportunities of cancer prevention1. In a recent Swiss study, there was a 25-50% response rate to invitations to cascade testing and at least one-in-three individuals at risk did not undergo testing. An index case possesses an average of 10 relatives eligible for testing, while the average rate of genetic tests per index case is only 1.51.
There are several barriers to cascade testing6,7. These include ineffective family communication of genetic risk information, low knowledge of cascade testing among index cases and primary care providers, and geographic barriers to receiving genetic services. Cascade testing uptake is also lower among male than female relatives and in distant compared to first-degree relatives. A facilitator of adherence to cascade testing is the parents’ desire to understand their children’s risk6. “Dear family” letters, digital chatbots (a technology-based simulated conversations), and direct contact programs have been shown to be effective in motivating cascade testing8.
Several initiatives exist to promote cascade testing. One such enterprise is the Cascade Resources Network, an independently run, non-profit organization that offers access to genetic testing, genetic counseling, variant interpretation, screening guidelines, and forums and support. It was developed by Memorial Sloan Kettering Cancer Center (MSK) fellows, Ryan Kahn and Sushmita Gordhandas. The network was created to increase the rate of genetic testing among relatives of patients with inherited cancer risk variants to help identify cancer early in families and, ultimately, to prevent future cancers. Similarly, the Swiss Cancer Genetic Predisposition Cascade Screening Consortium was established in 2016 to foster research related to the hereditary cancer predisposition. In particular, the Consortium promotes the CASCADE cohort, a family-based open-ended cohort targeting HBOC and LS variant-harboring families to elicit factors that enhance adherence to testing (NCT03124212).
Analyze genetic predisposition to cancer with the SOPHiA DDM™ Platform
Multi-gene testing is an efficient, affordable, and guideline-recommended9 approach to cascade testing as it allows for comprehensive assessment of biologically relevant hereditary cancer genes. The SOPHiA DDM™ Platform supports various next generation sequencing (NGS)-based Hereditary Cancer Applications to help clinician researchers characterize the complex mutational landscape associated with hereditary cancer disorders.
Powered by advanced analytics, users can detect challenging variants in a streamlined sample-to-report workflow, including:
Variant pathogenicity levels are assigned using machine learning complemented by guideline-driven ranking, helping to prioritize relevant variants and reduce interpretation time. Furthermore, deeper variant exploration is supported by Alamut™ Visual Plus, a full-genome browser that integrates numerous curated genomic and literature databases, guidelines, missense and splicing predictors.
To learn more about SOPHiA DDM™ for Hereditary Cancers, explore here or request a demo here.
References
1. Sarki M, et al. Cancers (Basel) 2022;14:1636.
2. Brown GR, et al. JAAPA 2020;33(12):10-16.
3. Mighton C, Lerner-Ellis JP. Genes Chromosomes Cancer 2022;61(6):356-381.
4. Dotson WD, et al. Clin Pharmacol Ther 2014;95(4):394-402.
5. Chen S, Parmigiani G. J Clin Oncol 2007;25(11):1329-1333.
6. Roberts MC, et al. Health Aff (Millwood) 2018;37(5):801-808.
7. O'Neill SC, et al. Hered Cancer Clin Pract 2021;19(1):40.
8. Campbell-Salome G, et al. (2022) Transl Behav Med 2022;12(7):800–809.
9. Daly MB, et al. J Natl Compr Canc Netw. 2021 Jan 6;19(1):77-102.
Companion diagnostics (CDx) are medical devices, specifically an in vitro diagnostic device (IVD), providing important information regarding the safe and effective use of therapeutics. The Food and Drug Administration (FDA) ascribes them three crucial functions: 1) identify patients more likely to benefit from a therapeutic; 2) determine patients at increased risk of serious side effects; and 3) monitor treatment responses for the purpose of adjusting dosage or regimens to improve safety and effectiveness. In our estimation, CDx act as a compass that directs the healthcare provider to the most appropriate treatment for each patient1.
The inception of CDx can be traced back to 1998, when the FDA granted concurrent approval for trastuzumab, a targeted cancer drug, and HercepTest™, a HER2 immunohistochemical assay. This milestone marked the birth of the drug-diagnostic co-development model, a transformative approach that has since witnessed consistent and substantial adoption2.
However, over the next 14 years, CDx advancement was slow, with the majority of new approvals occurring only in the past decade. In fact, from 1998 to 2012, approximately 20 new CDx were approved, whereas from 2013 to 2023, that number rose to 1343.
Today, approximately 50% of all new molecular entity (NME) approvals in oncology have an associated CDx or biomarker listed in the label required for safe and effective use (based on FDA approvals from 2021 and 2022 of NME in oncology3). Despite the historical tendency toward oncology products, applications in rare diseases and metabolic syndromes are evolving, paving the way for CDx to become an intrinsic part of precision medicine clinical trials across many indications.
The use of a companion diagnostics strategy in clinical trials, which we define here as using one or more biomarkers to pre-select and enroll patients more likely to respond to the experimental therapy, is commonly employed in oncology. In these trials, identification and pre-selection have significant advantages, allowing smaller patient groups to power the statistical analysis, potentially reducing overall costs, and increasing the likelihood of approval4.
But while a CDx strategy makes regulatory approval of a cancer drug more likely, it can simultaneously add complexity to the process:
Despite the evident complexity, the widespread adoption of next-generation sequencing (NGS) has made using companion diagnostics and deploying biomarker-driven strategies in clinical trials easier by permiting screening for multiple biomarkers simultaneously. Rather than rely on a one-biomarker-one-test model, NGS permits patients to be screened for eligibility for multiple therapeutics or clinical trials.. Moreover, targeted gene panels and broader approaches, such as comprehensive genomic profiling (CGP) and whole exome sequencing (WES) have further streamlined the development of multi-biomarker-driven CDx.
Returning to our analogy, while CDx acts as the compass, NGS technologies are the roadmap to determine the most suitable treatment for each patient.
There are two well-defined pathways to approval for companion diagnostics:
HercepTest™ followed the first pathway where the physical kit, produced by an IVD manufacturer, received a Pre-market Approval (PMA). In contrast, the precedent for the single-site model was only established much later with the publication of clinical evidence supporting the hypothesis that BRCA-mutated patients were more likely to benefit from treatment with olaparib—a PARP inhibitor first approved for the treatment of advanced ovarian cancer5,6.
Requiring a complex workflow and expert oversight, BRCA analysis was more conducive to the simplicity of the single-site model since the very nature of this pathway streamlines validation – validating a workflow within a single lab is less time-consuming than across multiple labs. This new approach played a pivotal role in rapidly advancing the commercial adoption of NGS applications.
Today, most NGS-powered CDx assays follow the single-site pathway3, creating a new set of challenges. Despite the increased simplicity, this model confines assays to single locations, limiting the capacity for sample analysis, significantly increasing turnaround times, and reducing patient access. In the new age of precision medicine, these limitations are being addressed through a decentralized testing and analysis model supported by technology-agnostic and easy-to-implement workflows.
While direct co-development of a CDx and therapeutic stands as the preferred regulatory model by the FDA, alternative approaches may be utilized due to the inherent challenges of aligning IVD and drug development.
While the development of a CDx and therapeutic are tightly entwined, drug developers and IVD manufacturers remain separate entities with a few exceptions. This requires the carefully selection of partner(s) within the IVD and CDx ecosystem to ensure successful programs.
Many questions must be addressed early in the process, as even suboptimal approaches can significantly delay and impact commercial uptake:
The advent of NGS technologies has heralded a healthcare revolution, propelling us toward data-driven precision medicine. Yet, as we push ahead in developing biomarker-driven applications for a plethora of indications, we face the potential for increased implementation challenges, threatening to complicate the patient journey.
The adoption of a decentralized, globally accessible, intuitive, and technology-agnostic SOPHiA DDM™ Platform, leveraging proprietary algorithms and a vast portfolio of robust NGS-based applications, is well positioned to streamline CDx co-development and implementation, enhancing access to analytically robust solutions without overtaxing healthcare resources.
Our holistic approach to co-development is poised to chart a course toward a more integrated future, arming developers with the necessary data and insights to tackle potential hurdles and maximize the time and resources allocated to clinical research programs.
At SOPHiA GENETICS, our unwavering commitment is to collaboratively engineer deployable solutions that elevate implementation and accessibility in precision testing while streamlining the process of analysis and interpretation. Explore the possibilities of SOPHiA DDM™ for BioPharma by visiting our dedicated page.
References
1 Food and Drug Administration (FDA). In Vitro Companion Diagnostic Devices: Guidance for Industry and Food and Drug Administration Staff. Issued on: August, 2014. Accessed on: October, 2023. Retrieved from: https://www.fda.gov/media/81309/download
2 Jørgensen JT. Drug-diagnostics co-development in oncology. Front Oncol. 2014;4:208. doi: 10.3389/fonc.2014.00208
3 FDA. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). Accessed on: October 2023. Retrieved from: https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools
4 Biotechnology Industry Organization (BIO). Clinical Development Success Rates and Contributing Factors 2011–2020. Accessed on: October 2023. Retrieved from: https://go.bio.org/rs/490-EHZ-999/images/ClinicalDevelopmentSuccessRates2011_2020.pdf
5 Ledermann J, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852-61. doi: 10.1016/S1470-2045(14)70228-1.
6 Deeks ED. Olaparib: first global approval. Drugs. 2015;75(2):231-240. doi: 10.1007/s40265-015-0345-6
7 Hanna TP, et al. Mortality due to cancer treatment delay: systematic review and meta-analysis. BMJ. 2020 Nov 4;371:m4087. doi: 10.1136/bmj.m4087
Genomic instability is a hallmark of cancer and targeting its mechanisms has helped inform effective therapeutic strategies1,2. However, there are limitations with current methods of genomic instability assessment. Here, we explore genomic instability in the context of homologous recombination deficiency and the value of deep learning-based methods of detection.
The DNA in our cells endure up to one million damaging events each day, caused by both exogenous (i.e. environmental) and endogenous (i.e. internal metabolic) factors3. These events activate a complex network of DNA damage response (DDR) pathways, which facilitate DNA repair and maintain the stability of the genome4. Mutations and/or dysfunction in DDR pathways can lead to unrepaired DNA damage, resulting in genomic instability4. Genomic instability, in turn, increases the cell’s propensity for genetic alterations that cause cancer initiation and progression4,5.
One of the major DDR pathways is the homologous recombination repair (HRR) pathway, responsible for repairing double-strand breaks (DSBs) in DNA5. Loss of function in HRR, known as homologous recombination deficiency (HRD), causes cells to rely on error-prone DNA repair pathways, resulting in the accumulation of genetic aberrations that lead to genomic instability5 (Fig 1). HRD is a well-established prognostic and predictive biomarker in different cancer types (e.g. ovarian, breast, prostate, and pancreatic)5–7.
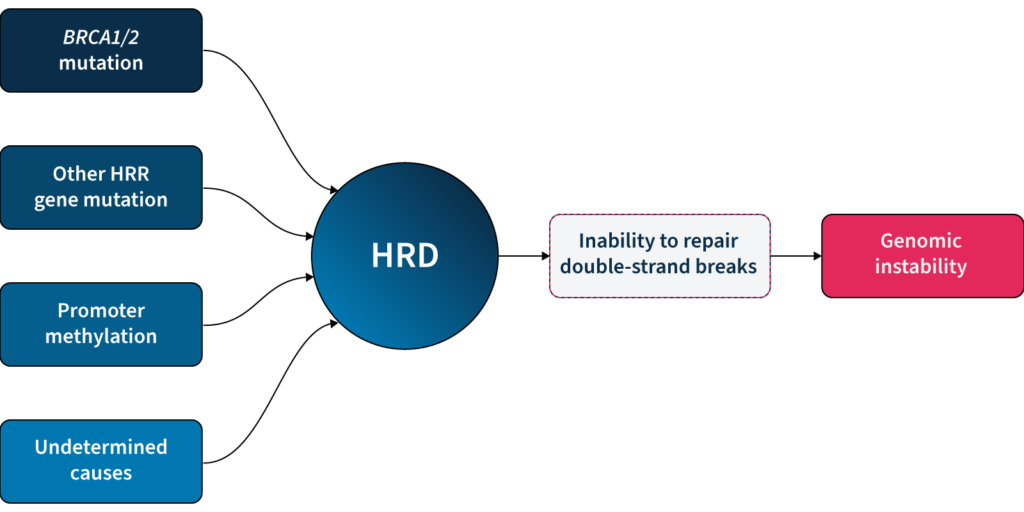
HRD-positive tumors are sensitive to targeted inhibition of poly-ADP ribose polymerase (PARP), key proteins involved in DSB repair7. By blocking PARP, the HRD-positive cell can no longer rely on error-prone pathways for DSB repair and the cell dies, a process known as ‘synthetic lethality’5,6. PARPi therapy has revolutionized the management of HRD-positive patients with advanced ovarian cancer, significantly improving progression-free survival when used as a first-line maintenance therapy8. PARPi therapies also have approved indications in breast, pancreatic and prostate cancer9, with trials underway in other cancer types, such as colorectal10.
Based on the predictive value of HRD status for PARPi benefit, clinical guidelines recommend HRD testing in patients with advanced ovarian cancer7,11,15. HRD status can be determined by examining 1) the underlying causes of HRD, and 2) the effect of HRD, i.e. genomic instability5,7. The most well-known causes of HRD are loss-of-function mutations in HRR genes, including BRCA1 and BRCA25,7. However, loss-of-function in HRR genes is diverse amongst patients12, making patient stratification solely based on genotyping challenging. Also, approximately 30–40% of HRD cases are due to unknown causes13,14. Measuring genomic instability allows the assessment of HRD, regardless of its underlying etiology5,7.
Genomic instability status can help identify a sub-group of women who are BRCA wild-type but may still derive benefit from PARPi therapy15. By measuring genomic instability, clinicians and researchers can therefore go beyond HRR mutation detection and expand the potential benefit of PARPi in patients.
Many methods for measuring genomic instability rely on the identification of specific mutational signatures or genomic ‘scars’ associated with large-scale structural rearrangements in chromosomes. In HRD-positive cancers, the characteristic genomic scars are loss of heterozygosity (LOH), large-scale state transitions (LST), and telomeric-allelic imbalance (TAI)16–18.
Click the boxes below to learn more:

A cross-chromosomal event that results in loss of part of a gene or entire gene(s) and the surrounding chromosomal region.

Chromosomal breaks between adjacent regions of at least 10 Mb.
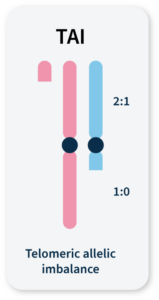
Accumulation of a discrepancy in the 1:1 allele ratio at the end of the chromosome (telomere).
The combined number of LOH, LST, and TAI events generate a genomic instability score (GIS) that reflects the level of genomic instability. Some commercially available HRD tests combine tumor BRCA mutation testing with a GIS5,19. Methods that integrate multiple genome-wide signatures (e.g. HRDetect) are among the most promising for detecting HRD status7,20. However, both GIS and HRDetect methods require deep genomic profiling data (>30x coverage), which can be costly and difficult to implement in routine analysis.
Alternative approaches that rely on the detection of copy number changes from WGS at low (~1x) sequencing depth (low-pass WGS) can predict tumor HRD status21,22 and provide an affordable and easy-to-implement HRD detection method. However, the sensitivity of existing methods that solely rely on this type of genomic scar to identify HRD samples is limited, and their utility in a clinical context remains untested22.
Unlocking the full potential of low-pass WGS in HRD detection requires going beyond the enumeration of biomarker events and examining alternative features of the cell that can result from genomic instability.
To overcome the limitations of current genomic instability measures, our expert team at SOPHiA GENETICS developed the GIInger™ algorithm exclusively available on the SOPHiA DDM™ Platform. GIInger™ is a deep learning-based approach to measuring genomic instability in ovarian cancer samples. Rather than relying on the enumeration of biomarkers events, GIInger™ leverages differences in the spatial distribution of genomic scars in low-pass WGS coverage profiles23.
Let’s take a closer look at how the algorithm predicts genomic instability status (Fig 2):
Unfamiliar with deep learning terminology? Read our guide on machine learning jargon.
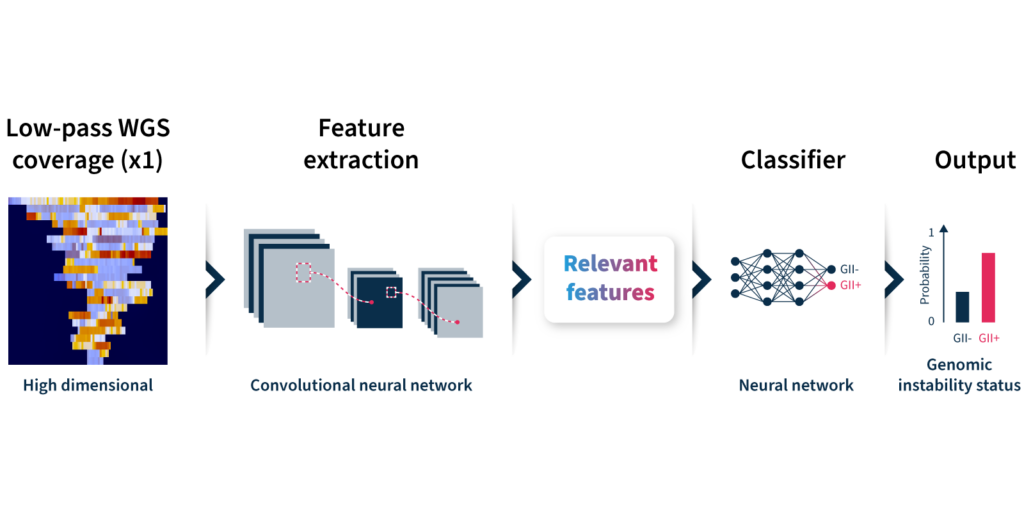
By adopting GIInger™ into next generation sequencing (NGS) workflows, clinical researchers can benefit from an in-house, affordable approach to genomic instability measurement. The SOPHiA DDM™ Platform offers applications that enable laboratories to easily implement GIInger™ into their routine NGS analysis:
Want to see how GIInger™ can help maximize insights from your data? Get in touch with our team and request a demo.
SOPHiA GENETICS products are for Research Use Only and not for use in diagnostic procedures unless specified otherwise.
SOPHiA DDM™ Dx Hereditary Cancer Solution, SOPHiA DDM™ Dx RNAtarget Oncology Solution and SOPHiA DDM™ Dx Homologous Recombination Deficiency Solution are available as CE-IVD products for In Vitro Diagnostic Use in the European Economic Area (EEA), the United Kingdom and Switzerland. SOPHiA DDM™ Dx Myeloid Solution and SOPHiA DDM™ Dx Solid Tumor Solution are available as CE-IVD products for In Vitro Diagnostic Use in the EEA, the United Kingdom, Switzerland, and Israel. Information about products that may or may not be available in different countries and if applicable, may or may not have received approval or market clearance by a governmental regulatory body for different indications for use. Please contact us to obtain the appropriate product information for your country of residence.
All third-party trademarks listed by SOPHiA GENETICS remain the property of their respective owners. Unless specifically identified as such, SOPHiA GENETICS’ use of third-party trademarks does not indicate any relationship, sponsorship, or endorsement between SOPHiA GENETICS and the owners of these trademarks. Any references by SOPHiA GENETICS to third-party trademarks is to identify the corresponding third-party goods and/or services and shall be considered nominative fair use under the trademark law.