The discovery of mitochondria
Within every cell in the human body, there are thousands of mitochondria. They are, for all intents and purposes, little proto-bacteria subsumed into your cells, providing them with energy in an ancient symbiotic relationship — the cell sheltering the mitochondria, and the mitochondria producing energy for the cell by performing aerobic respiration. Each mitochondrion has its own tiny circular genomes , which means that instead of just a solitary genome in the nucleus, each eukaryotic cell is studded with thousands of copies of micro-genomes (creating “heteroplasmy”, where one cell can contain multiple non-identical genomes). One of the earliest descriptions of mitochondria came in 1890, when Richard Altmann called them “bioblasts” or “life germs”.1,2 The description of the first mitochondria-based disease came 70 years later in 1962, based partly on analysis of a hyperthyroid patient whose tissue displayed physically abnormal mitochondria and had biochemically “loose coupling” of aerobic respiration.3
The early landmark work on mitochondrial genomes started arriving around the late 1970s and early 1980s. The mitochondrial genome was in fact the first complete eukaryotic genome ever sequenced. This was in a pre-PCR, Sanger-sequencing world when the idea of even thermal cycling hadn’t taken hold. The first paper that described the mitochondrial genome’s unusual properties is an interesting time capsule for how laborious molecular biology and genome sequencing was 40 years ago (this is actually a separate paper from the one describing the complete mitochondrial genome, which came a few years later in 1981).4,5 Upwards of 80 proteins participate in oxidative phosphorylation (one of the component steps of aerobic respiration), with the mitochondrial genome encoding 13 of them.6 Though the exact percentage of mitochondria inherited from either parent is debatable, the vast, vast majority is inherited from the mother, and so maternal mitochondrial genetics will dominate the child’s mitochondrial genetics.7
Disease-causing mutations in mitochondrial DNA
Mutations in the mitochondrial genome cause a litany of human diseases, affecting 1 in 5000 people.8,9 Mitochondrial disorders are particularly common, as the mitochondrial genome experiences mutations at a far higher rate (commonly thought to be between 10- and 100-fold higher) than the nuclear genome.10 This susceptibility to mutation is thought to arise from mitochondrial DNA’s (mtDNA’s) proximity to radical oxygen species produced during respiration. The symptoms of mitochondrial disorders are diverse, from neurological disorders like migraines, strokes, epilepsy, and dementia; damage to the senses; to essentially any organ you can think of, resulting in diseases as diverse as diabetes and thyroid disease, renal failure, heart failure, and disorders of speech. There have been demonstrated instances of patients with complex metabolic disorders, whose genetic etiology was left unexplained by nuclear sequencing and was only clarified when their samples were re-sequenced to examine their mitochondrial genomes.
Inherited mitochondrial disorders can occur in any tissue type. A small load of inherited mtDNA mutations tends not to create medical conditions in a person’s lifetime, though determining what the threshold is for inherited mutations that do cause disease is difficult to pin down, and likely varies by the tissue that a mutation occurs in. Mitochondrial disorders are not only inherited but can arise due to a previously unseen mutation, which can lead to a complicated diagnosis.
Mutations in the mitochondrial genome cause a litany of human diseases, affecting 1 in 5000 people
Early analysis of mtDNA
In the era before robotics, Sanger sequencing was a laborious task. The mitochondrial genome was cut into pieces using specific restriction enzymes, cloned into a plasmid, and sequenced using radioactively labeled primers. 16,569 base pairs were read and recorded through Sanger runs on slab gels,5 which are as thin and fragile as tissue paper and can accommodate 100-200 bases per run at the most. It was brute-force methods that both sequenced the entire mitochondrial genome and found its unusual properties, like its reassignment of the UGA stop codon to tryptophan and its unique suite of tRNA genes that don’t match the nuclear genome.
In the era before laboratory computers, sequence alignment, and genome browsers, mutations in mtDNA were detected by “sequence gazing”: just looking at sequences until you spotted differences. A missense mutation had to be found by hand, and a frameshift analyzed by eye and compared against the unique codon table of the mitochondria. The diagnostic yield for mitochondrial disorders in adults can be as low as 11% using Sanger sequencing, but as high as 40-60% using next generation sequencing (NGS).11
NGS analysis of mtDNA in the 21st century
Thankfully, the introduction of NGS has made both the lab work and analysis of mitochondrial genetics data much, much more approachable, no matter the size and background of the lab. NGS techniques make detecting uncommon mutations in large pools of data — like, say, a large and heterogeneous collection of mitochondrial genomes — a magnitude of order easier than Sanger sequencing, on top of being able to multiplex a variety of different samples in a single sequencing run. Rare, disease-causing mitochondrial genome mutations may be below the limit of detection of traditional Sanger sequencing, in some cases requiring that NGS be performed.
In human cells, there is only one nuclear genome, containing about 3 billion bases. The mitochondrial genome is only 16,659 base pairs long, but the varied number of mitochondrial genomes per cell, and the variation between each mitochondrion, makes the sequencing depth and coverage uniformity made possible by NGS critical. The relatively higher number of mitochondrial (versus nuclear) genome copies also contributes to increased mitochondrial DNA mutational rates — more copies, more chances for error. Depending on the sample type, there is huge variability in the number of mitochondria and thus the copy number of mitochondrial genomes. Less energy-intensive tissues like the lungs can have as few as several hundred mitochondria per cell, whereas tissues that require huge amounts of energy, like the liver, heart, and skeletal muscle, can have 1000-7000 mitochondria per cell.12 Furthermore, the number of mitochondria per cell can itself be a marker for disease (called mitochondrial DNA depletion syndromes), though this can be detected using quantitative PCR, without the need for a sequencing run.
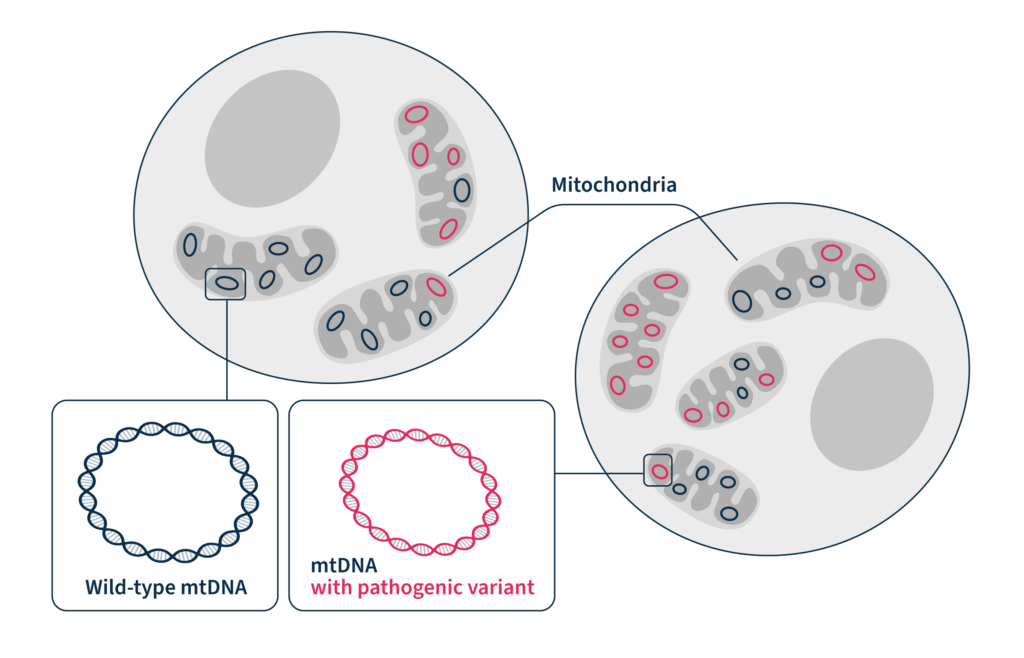
Heteroplasmy, the variation in abundance of pathogenic variants due to the fact that there are thousands of distinct mitochondrial genomes per cell, can make analysis difficult. On top of that, nuclear mitochondrial sequences (NUMTs), translocations of mitochondrial DNA to the nucleus, can make capturing data specific to the mitochondrial genome more complex than simple whole-cell sequencing. Finally, even if a genetic test is ordered in the clinic, these normally do not include the mitochondrial genome, thus completely discounting mitochondrial disorders from the outset.
Characterization of mitochondrial disorders with SOPHiA DDM™ and Alamut™ Visual Plus
At SOPHiA GENETICS, we offer integrated mitochondrial testing as part of our clinical exome and whole exome solutions, enabling the identification of pathogenic variants in the human exome and mitochondrial genome in a single experiment. Our panel design and the sophisticated algorithms used by the SOPHiA DDM™ platform address the unique challenges associated with the mitochondrial genome, such as the high and variable amount of mtDNA and heteroplasmy, to provide coverage uniformity and accurately and confidently identify variants in mtDNA in a streamlined analytical workflow.
Software like Alamut™ Visual Plus can take the eyestrain out of sequence gazing by allowing visualization of detected variants in a comprehensive full genome browser. SOPHiA DDM™ and Alamut™ Visual Plus can simplify the complex and time-consuming assessment of mitochondrial variant pathogenicity by pre-classifying and annotating variants with curated, up-to-date information from literature sources such as PubMed® and MasterMind®, and genomic databases such as ClinVar, OMIM®, and dbSNP, in a single, user-friendly environment.
Where sequencing and gazing at a mitochondrial DNA sequence in the 1980s could have taken weeks of squinting at gels, the SOPHiA DDM™ platform turns around sequencing data in a matter of hours, and Alamut™ Visual Plus can accelerate variant interpretation to a few clicks.

References
- Altmann R. Verlag von Veit & Comp, Leipzig 1890. https://www.deutschestextarchiv.de/book/view/altmann_elementarorganismen_1890.
- O’Rourke B. Front Physiol 2010;1:7.
- Luft R, Ikkos D, Palmieri G, et al. J Clin Invest 1962;41:1776–1804.
- Barrell B, Bankier AT, Drouin J. Nature 1979;282:189–94.
- Anderson S, Bankier AT, Barrell BG, et al. Nature 1981;290:457–65.
- Sylvester JE, Fischel-Ghodsian N, Mougey EB, et al. Genet Med 2004;6:73–80.
- Sato M, Sato K. Biochim Biophys Acta 2013;1833:1979–84.
- Taylor RW, Turnbull DM. Nat Rev Genet 2005;6:389–402.
- Falk MJ, Sondheimer N. Curr Opin Pediatr 2010;22:711–16.
- Ludwig LS, Lareau CA, Ulirsch JC, et al. Cell 2019;176:1325-1339.e22.
- Wortmann SB, Koolen DA, Smeitink JA, et al. J Inherit Metab Dis 2015;38:437–43.
- D’Erchia AM, Atlante A, Gadaleta G, et al. Mitochondrion 2015;20:13–21.